| Sections | Required Section | Optional Section | Output options | Standby screen | Progress | Output files |
| Formats | FASTA format | GFF3 format | GenBank flat file format | |||
| Other | Process details | References |
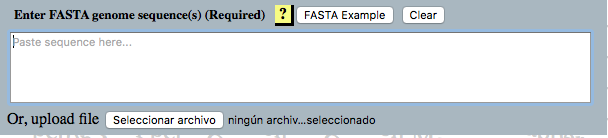
Required Section
Paste a Fasta example sequence clicking here. ↓
Optional Section
paste them here (or upload a file) to speed up the process.
Both GFF and GenBank accepted.
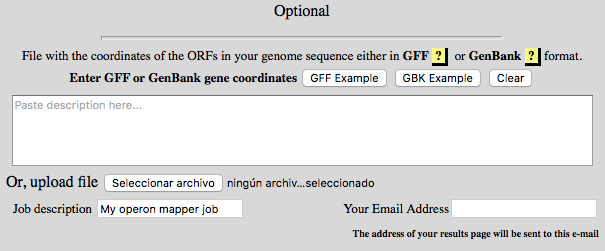
| Aditionally in this section you ↑ can create an identification name to your job, that can be usefull if you send several sequences. | If you provide an ↑ email address, you will be notified as soon as your job is finished. |
Output options
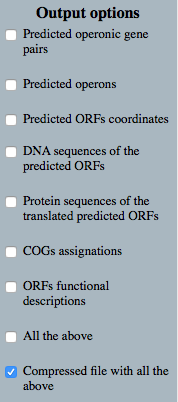
|
←Predicted operonic gene pairs with their corresponding confidence values of being part of the same operon ←List of operons with their conforming genes ←Predicted ORFs coordinates ←DNA sequences of the predicted ORFs ←Protein sequences of the translated predicted ORFs ←Orthology assignment of proteins to their corresponding COGs groups ←Proteins functional descriptions |
|
←Or select one of the all buttons:
|
Standby screen
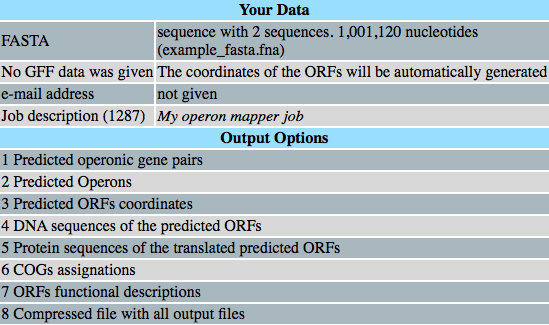
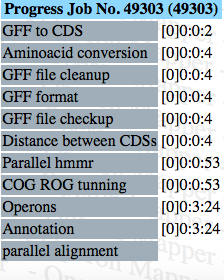
Progress
[D]H:M:S
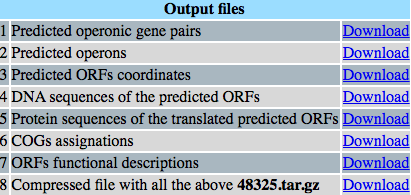
Output files
If you have selected the compressend option, it will be downloaded.
FASTA format
The format also allows for sequence names and comments to precede the sequences.
Example:
>Ecoli_K-12 | 0002 AGCTTTTCATTCTGACTGCAACGGGCAATATGTCTCTGTGTGGATTAAAAAAAGAGTGTCTGATAGCAGC TTCTGAACTGGTTACCTGCCGTGAGTAAATTAAAATTTTATTGACTTAGGTCACTAAATACTTTAACCAA TATAGGCATAGCGCACAGACAGATAAAAATTACAGAGTACACAACATCCATGAAACGCATTAGCACCACC >Ecoli_K-12 | 0006 ATTACCACCACCATCACCATTACCACAGGTAACGGTGCGGGCTGA >Ecoli_K-12 | 0042 CCCGCACCTGACAGTGCGGGCTTTTTTTTTCGACCAAAGGTAACGAGGTAACAACCATGCGAGTGTTGAA GTTCGGCGGTACATCAGTGGCAAATGCAGAACGTTTTCTGCGTGTTGCCGATATTCTGGAAAGCAATGCC AGGCAGGGGCAGGTGGCCACCGTCCTCTCTGCCCCCGCCAAAATCACCAACCA
GFF3 format
(based on http://gmod.org/wiki/GFF3)
GFF3 format is a flat tab-delimited file.
The first line of the file is a comment that identifies the file format and version.
This is followed by a series of data lines, each one of which corresponds to an annotation.
Here is a miniature GFF3 file:
##gff-version 3 bsu:NC_000964 Ric CDS 410 1750 . + 0 ID=BSU00010; bsu:NC_000964 Ric CDS 1939 3075 . + 0 ID=BSU00020; bsu:NC_000964 Ric CDS 3206 3421 . + 0 ID=BSU00030; bsu:NC_000964 Ric CDS 3437 4549 . + 0 ID=BSU00040; bsu:NC_000964 Ric CDS 4567 4812 . + 0 ID=BSU00050; bsu:NC_000964 Ric CDS 4867 6783 . + 0 ID=BSU00060; bsu:NC_000964 Ric CDS 6994 9459 . + 0 ID=BSU00070; bsu:NC_000964 Ric CDS 14847 15794 . - 0 ID=BSU00080; bsu:NC_000964 Ric CDS 15915 17381 . + 0 ID=BSU00090;The ##gff-version 3 line is required and must be the first line of the file. It introduces the annotation section of the file. The 9 columns of the annotation section are as follows:
Column 1: "seqid"
The ID of the landmark used to establish the coordinate system for the current feature. IDs may contain any characters, but must escape any characters not in the set [a-zA-Z0-9.:^*$@!+_?-|]. In particular, IDs may not contain unescaped whitespace and must not begin with an unescaped ">". To escape a character in this, or any of the other GFF3 fields, replace it with the percent sign followed by its hexadecimal representation. For example, ">" becomes "%E3". See URL Encoding (or: 'What are those "%20" codes in URLs?') for details.
Column 2: "source"
The source is a free text qualifier intended to describe the algorithm or operating procedure that generated this feature. Typically this is the name of a piece of software, such as "Genescan" or a database name, such as "Genbank." In effect, the source is used to extend the feature ontology by adding a qualifier to the type creating a new composite type that is a subclass of the type in the type column. It is not necessary to specify a source. If there is no source, put a "." (a period) in this field.
Column 3: "type"
The type of the feature (previously called the "method"). This is constrained to be exactly CDS. Records with any other value will be ignored.
Columns 4 & 5: "start" and "end"
The start and end of the feature, in 1-based integer coordinates, relative to the landmark given in column 1. Start is always less than or equal to end. For zero-length features, such as insertion sites, start equals end and the implied site is to the right of the indicated base in the direction of the landmark. These fields are required.
Column 6: "score"
The score of the feature, a floating point number. As in earlier versions of the format, the semantics of the score are ill-defined. It is strongly recommended that E-values be used for sequence similarity features, and that P-values be used for ab initio gene prediction features. If there is no score, put a "." (a period) in this field.
Column 7: "strand"
The strand of the feature. + for positive strand (relative to the landmark), - for minus strand, and . for features that are not stranded. In addition, ? can be used for features whose strandedness is relevant, but unknown.
Column 8: "phase"
For features of type "CDS", the phase indicates where the feature begins with reference to the reading frame.
The phase is one of the integers 0, 1, or 2, indicating the number of bases that should be
removed from the beginning of this feature to reach the first base of the next codon.
In other words,
a phase of "0" indicates that the next codon begins at the first base of the region described by the
current line, a phase of "1" indicates that the next codon begins at the second base of this region,
and a phase of "2" indicates that the codon begins at the third base of this region.
This is NOT to be confused with the frame, which is simply start modulo 3. If there is no phase, put a "." (a period) in this field.
For forward strand features, phase is counted from the start field. For reverse strand features, phase is counted from the end field.
The phase is required for all CDS features.
Column 9: "attributes"
A list of feature attributes in the format tag=value. Multiple tag=value pairs are separated by semicolons. URL escaping rules are used for tags or values containing the following characters: ",=;". Spaces are allowed in this field, but tabs must be replaced with the %09 URL escape. This field is not required.
Column 9 Tags
Column 9 tags have predefined meanings:
ID
Indicates the unique identifier of the feature. IDs must be unique within the scope of the GFF file.
Name
Display name for the feature. This is the name to be displayed to the user. Unlike IDs, there is no requirement that the Name be unique within the file.
Alias
A secondary name for the feature. It is suggested that this tag be used whenever a secondary identifier for the feature is needed, such as locus names and accession numbers. Unlike ID, there is no requirement that Alias be unique within the file.
Parent
Indicates the parent of the feature. A parent ID can be used to group exons into transcripts, transcripts into genes, and so forth. A feature may have multiple parents. Parent can *only* be used to indicate a partof relationship.
Target
Indicates the target of a nucleotide-to-nucleotide or protein-to-nucleotide alignment. The format of the value is "target_id start end [strand]", where strand is optional and may be "+" or "-". If the target_id contains spaces, they must be escaped as hex escape %20.
Gap
The alignment of the feature to the target if the two are not collinear (e.g. contain gaps). The alignment format is taken from the CIGAR format described in the Exonerate documentation. http://cvsweb.sanger.ac.uk/cgi-bin/cvsweb.cgi/exonerate?cvsroot=Ensembl). See the GFF3 specification for more information.
Derives_from
Used to disambiguate the relationship between one feature and another when the relationship is a temporal one rather than a purely structural "part of" one. This is needed for polycistronic genes. See the GFF3 specification for more information.
Note
A free text note.
Dbxref
A database cross reference. See the GFF3 specification for more information.
Ontology_term
A cross reference to an ontology term. See the GFF3 specification for more information.
Multiple attributes of the same type are indicated by separating the values with the comma "," character, as in: Parent=AF2312,AB2812,abc-3 Note that attribute names are case sensitive. "Parent" is not the same as "parent". All attributes that begin with an uppercase letter are reserved for later use. Attributes that begin with a lowercase letter can be used freely by applications. You can stash any semi-structured data into the database by using one or more unreserved (lowercase) tags.
GenBank flat file format
The GenBank format is formed by many data elements (see Sample GenBank Record for a detailed description of each data element)
For our purposes, only LOCUS, DEFINITION, VERSION, FEATURES and CDS are needed. Is VERY IMPORTANT that the first field after VERSION element match exactly with the FASTA identificators in the sequence file, since that is the engaging element. The CDS or Coding Sequence especifies region of nucleotides that corresponds with the sequence of amino acids in a protein (location includes start and stop codons).
The CDS feature may include an amino acid translation as well as other data such as note, codon_start, function, product, protein_id, db_xref, etc., but are not used.
Here is a "minimum" example:
LOCUS Ecoli_K-12 1 bp DNA UNA 01-Dec-2016
DEFINITION Ecoli_K-12_MG1655_Example
VERSION Ecoli_K-12
FEATURES Location/Qualifiers
CDS 337..2799
/gene="orf00002"
CDS 2801..3733
/gene="orf00003"
CDS 3734..5020
/gene="orf00005"
Process details
1) Data acquisition.
This procedure is performed using a web page written in HTML and JavaScript. The only required input for the operon prediction process is the genome nucleotide sequence, nevertheless, the ORFs genomic coordinates can also be provided by the user, either in GFF (General Feature Format) or GenBank format.
2) Sequence analyses.
The analysis is divided in five different tasks: 2.1) ORFs prediction using the Prodigal software that employs dynamic programming to accurately predicts
the 5' and 3' ends of all ORFs in the given nucleotide sequence (7).
2.2) Orthology genes assignments, which are determined considering the most significant E-value of Hidden Markov Models (HMMs) searches using the hmmsearch program (8). This HMMs search process employs a set of models that represents each of the 4,873 COGs (Clusters of Orthologous Groups of proteins) (9) and 8,539 ROGs (1) groups.
2.3) Intergenic distances evaluation, which is determined based on the ORFs' coordinates using a homemade Perl program.
2.4) Operon prediction, which is done by using an artificial neural net-work implemented in R. The inputs of our network to predict whether two contiguous genes are part of the same operon are the intergenic distance between them and a score value that reflects the functional relationship of their corresponding protein products. These scoring values have been defined in the STRING database (12) for different pairs of proteins according to the COG or ROG group of orthology to which they belong. This step is the core process of Operon-mapper where a confidence value of a pair of genes to be part of the same operon is evaluated. This confidence value is normalized between 0 and 1. A value greater than 0.5 indicates that a gene pair belongs to the same operon. The greatest accuracies for confidence values are those near to 0 or near to 1, and the lowest accuracies are those near to 0.5.
2.5) Gene function assignments, which are based on the most significant BLAST (11) against a core set of well characterized proteins from the Uniprot Knowledgebase consisting of non-fragmented proteins that are from Bacteria or Archaea. These proteins have an evidence level 2 or lower, corresponding to protein which existence are supported at tran-scriptional and proteomics level (13). The objective of this analysis is to have a first and reliable annotation for each predicted ORF as rapidly as possible.
3. Results delivery.
A Perl program is used to build an HTML page where a user has the opportunity to easily choice the file or set of files with the results of the different analyses performed by Operon-mapper including: i) Predicted operonic gene pairs with their corresponding confidence values of being part of the same operon, ii) List of operons with their conforming genes, iii) Predicted ORFs coordinates, iv) DNA sequences of the predicted ORFs, v) Protein sequences of the translated predicted ORFs, vi) Orthology assignment of proteins to their corre-sponding COG or ROG groups, v) Proteins functional descriptions, vi) All the above outflies, and vii) A compressed file with all the above outflies.
References
- 1. Taboada,B., Verde,C. and Merino,E. (2010) High accuracy operon prediction method based on STRING database scores. Nucleic Acids Res., 38.
- 2. Zaidi,S.S.A. and Zhang,X. (2016) Computational operon prediction in whole-genomes and metagenomes. Brief. Funct. Genomics, 10.1093/bfgp/elw034.
- 3. Pertea,M., Ayanbule,K., Smedinghoff,M. and Salzberg,S.L. (2009) OperonDB: A comprehensive database of predicted operons in microbial genomes. Nucleic Acids Res., 37.
- 4. Okuda,S. and Yoshizawa,A.C. (2011) ODB: A database for operon organizations, 2011 update. Nucleic Acids Res., 39.
- 5. Mao,F., Dam,P., Chou,J., Olman,V. and Xu,Y. (2009) DOOR: A database for prokaryotic operons. Nucleic Acids Res., 37.
- 6. Taboada,B., Ciria,R., Martinez-Guerrero,C.E. and Merino,E. (2012) ProOpDB: Prokaryotic Operon DataBase. Nucleic Acids Res., 40, D627-31.
- 7. Hyatt,D., Chen,G.-L., Locascio,P.F., Land,M.L., Larimer,F.W. and Hauser,L.J. (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics, 11, 119.
- 8. Eddy,S.R. (2011) Accelerated Profile HMM Searches. PLoS Comput. Biol., 7, e1002195.
- 9. Tatusov,R.L., Fedorova,N.D., Jackson,J.D., Jacobs,A.R., Kiryutin,B., Koonin,E. V, Krylov,D.M., Mazumder,R., Mekhedov,S.L., Nikolskaya,A.N., et al. (2003) The COG database: an updated version includes eukaryotes. BMC Bioinformatics, 4, 41.
- 10. Chang,C. and Lin,C. (2011) LIBSVM : A Library for Support Vector Machines. Science (80-. )., 10.1145/1961189.1961199.
- 11. Altschul,S.F., Madden,T.L., Schäffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res., 25, 3389 - 3402.
- 12. Jensen,L.J., Kuhn,M., Stark,M., Chaffron,S., Creevey,C., Muller,J., Doerks,T., Julien,P., Roth,A., Simonovic,M., et al. (2009) STRING 8 - A global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res., 37.
- 13. Apweiler,R., Bairoch,A., Wu,C.H., Barker,W.C., Boeckmann,B., Ferro,S., Gasteiger,E., Huang,H., Lopez,R., Magrane,M., et al. (2004) UniProt: the Universal Protein knowledgebase. Nucleic Acids Res., 32, D115-9.
- 14. Salgado,H., Peralta-Gil,M., Gama-Castro,S., Santos-Zavaleta,A., Muñiz-Rascado,L., García-Sotelo,J.S., Weiss,V., Solano-Lira,H., Martínez-Flores,I., Medina-Rivera,A., et al. (2013) RegulonDB v8.0: Omics data sets, evolutionary conservation, regulatory phrases, cross-validated gold standards and more. Nucleic Acids Res., 41.
- 15. Sierro,N., Makita,Y., De hoon,M. and Nakai,K. (2008) DBTBS: A database of transcriptional regulation in Bacillus subtilis containing upstream intergenic conservation information. Nucleic Acids Res., 36.